Familiäre Hypercholesterinämie
Begutachtet von Dr Krishna Vakharia, MRCGPZuletzt aktualisiert von Dr Colin Tidy, MRCGPZuletzt aktualisiert 15. Juni 2023
Erfüllt die Anforderungen des Patienten Richtlinien des Patienten
- HerunterladenHerunterladen
- Teilen
- Language
- Diskussion
- Audio-Version
- Zu bevorzugten Quellen bei Google hinzufügen
Familial hypercholesterolaemia is inherited high cholesterol. It is a condition where you have a very high cholesterol level in your blood.
Auf einen Blick
Familial hypercholesterolaemia (FH) means inherited high cholesterol levels in your blood.
You won't know if you have FH without a blood test; symptoms like fatty deposits appear later.
FH increases your risk of early heart disease, stroke, and peripheral artery disease.
Diagnosis involves blood tests, checks for fatty deposits, family history, and genetic testing.
Treatment includes medication, a healthy lifestyle, and sometimes specialised procedures.
Early treatment can help people with FH have a normal life expectancy.
What is familial hypercholesterolaemia (FH)?
Familial hypercholesterolaemia (FH) is called familial because it runs in the family (the other word for this is 'inherited'). Hypercholesterolaemia means cholesterol levels in your blood are too high. Too much cholesterol in your blood causes cholesterol deposits in your arteries and so an increased risk of early Herzerkrankungen (coronary heart disease), Schlaganfall und periphere Arterienerkrankung. See also the leaflet on Kardiovaskuläre Erkrankung (Atherom).
To find out more about the effect raised cholesterol has on your risk of heart attack and stroke, see the separate leaflet called High Cholesterol. Cholesterol levels in your blood are normally controlled when cholesterol is taken back to your liver and broken down.
In FH, you inherit at least one faulty gene, which means your liver is much less able to remove excess LDL cholesterol (sometimes referred to as 'bad cholesterol' or non-HDL cholesterol) from your blood. HDL cholesterol ('good cholesterol') helps clear the cholesterol out of your arteries.
What genes are involved in familial hypercholesterolaemia (FH)?
If you inherit a faulty gene from one parent, it is called 'heterozygous FH'. Without treatment, people with heterozygous FH are at higher risk of heart attack than average, and may have a heart attack in their 40s or 50s even if they have no other risk factors for heart disease.
In rare cases, you can inherit a copy of the faulty gene from both parents. This is called 'homozygous FH'. People with homozygous FH have exceptionally high levels of cholesterol - usually 10-20 mmol/L and sometimes even higher. Without treatment, people with homozygous FH can have heart attacks in their 20s.
There are three main genes that can be involved in FH:
LDL receptor genes: this is the most common faulty gene in FH. It means there aren't enough LDL receptors, so cholesterol builds up in the blood.
APOB gene (apolipoprotein B): a fault with this gene means LDL cholesterol can’t bind well to LDL receptors so that blood levels of LDL remain high.
PCSK9 gene: a fault with this gene causes LDL receptors to be broken down in the liver so they can’t remove cholesterol out of your blood stream.
How common is familial hypercholesterolaemia (FH)?
Heterozygous FH affects between 1 in 250 and 1 in 500 people. However homozygous FH is very rare and affects about 1 in a million people.
Signs and symptoms of familial hypercholesterolaemia (FH)
Ein hoher Cholesterinspiegel level doesn't make you feel ill. You won't know you have it without having a blood test.
The most important feature is the development of heart disease at a young age. This is caused by patches (plaques) of atheroma developing within the walls of the heart (coronary) arteries (see below). This can lead to a heart attack (myocardial infarction) as a young adult. You might also notice:
Xanthome - fatty cholesterol-rich deposits in the skin, usually found around the elbows, knees, buttocks and tendons.
Xanthelasma - fatty deposits in the eyelids.
Arcus senilis - a white ring around the cornea (the coloured part of the eye).
Diagnosing familial hypercholesterolaemia (FH)
You may be diagnosed by chance if you go for a health screening check or you may notice fatty deposits on the skin or around the eyes.
The levels of raised cholesterol your doctor will be looking for are:
Total cholesterol over 7.5 mmol/L in an adult (6.7 mmol/L in a young person under 16 years); und/oder
LDL-cholesterol above 4.9 mmol/L in an adult (4.0 mmol/L in a young person under 16 years).
Levels of cholesterol in your blood vary over time, so your doctor may want to do two tests to confirm that you have high cholesterol.
Your doctor should be able to make a definite diagnosis of FH if you have this level of raised cholesterol as well as the fatty deposits on your tendons (described above). You can also be diagnosed if you have genetic testing which proves you have a copy of the faulty gene found in FH.
They may suspect FH if you have this level of raised cholesterol and a family history of early coronary heart disease:
If you or a 'first-degree relative' - parent, brother, sister or your child - have a heart attack under the age of 60; oder
If a 'second-degree relative' - uncle, aunt, nephew, niece, grandparent - has a heart attack under the age of 50; oder
If other close relatives have cholesterol raised to the levels above.
This photo shows the typical signs of high cholesterol around the eyes - these pale deposits are called xanthelasma:
Xanthelasma
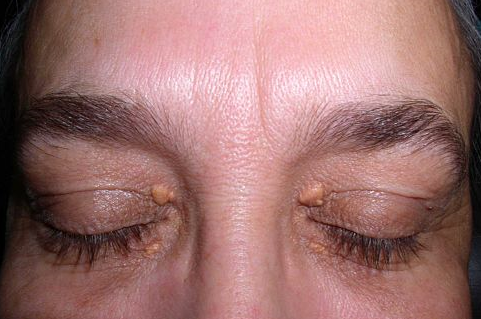
© Von Klaus D. Peter, CC-BY-3.0, über Wikimedia Commons
Testing for familial hypercholesterolaemia (FH)
If you have suspected or definite FH, you should be referred to a specialist FH centre for DNA testing to confirm the diagnosis.
If this testing confirms that you have FH, other members of your family should also be referred for DNA testing to see if they have FH as well. This includes all first-degree (parents, brothers and sisters, children) and second-degree (uncles aunts, nephews, nieces, grandparents, grandchildren) relatives. This 'cascade testing' should be carried out in a specialist centre.
Cascade testing is very important because it can allow other members of your family who have FH to be diagnosed and treated. This can prevent them having heart attacks at an early age.
Treating familial hypercholesterolaemia (FH)
Being a genetic disorder, FH is not caused by an unhealthy lifestyle. However, keeping yourself in the best physical condition will help to prevent future problems. Things you can do to help yourself include:
Gesunde Ernährung: in general, it's a good idea to eat healthily. But it's important to note that what you eat makes hardly any difference to your cholesterol level.
Getting a reasonable amount of exercise. Ideally this should be at least 30 minutes of moderate-intensity or high-intensity physical exercise at least five times a week.
Avoiding smoking: this is vital if you have high cholesterol, but even more if you have FH.
Maintaining a normal weight: but slim people can have high cholesterol too. Being overweight doesn't cause high cholesterol.
You will be offered medication to help bring your cholesterol level down - you will need to take this medicine for life. The usual medicine to start with is a Statin. The most common statin used is called atorvastatin. The aim will be to reduce your LDL cholesterol by at least 50% compared to your pre-treatment level.
Another medicine called Ezetimib is sometimes given:
Instead of a statin, if there is a medical reason which means you cannot take a statin.
Instead of a statin, if you have side-effects which prevent you from taking a statin.
In addition to a statin, if your cholesterol does not come down by enough on statin treatment.
Treatment for homozygous FH
If you have homozygous FH, you may need other treatments, including LDL-lowering apheresis. This is a treatment which filters LDL cholesterol out of the blood. It is offered to those who have the greatest risk of developing problems. Occasionally you may be referred for apheresis if you have heterozygous FH and you develop heart disease despite maximum doses of lipid-lowering tablets.
Treatment for children with FH
Children with FH will usually need to start treatment by the age of 10 years. This is because fatty deposits in the arteries develop very young in people with FH. If other people in the family have developed heart disease at a very young age, children with FH may need to start treatment before the age of 10 years or take more than one drug to reduce their cholesterol.
Some children may need apheresis. - in particular, those with the rare homozygous FH where they have inherited a faulty gene from each parent.
Hinweis der Redaktion
Dr. Krishna Vakharia, 13. September 2024
The National Institute for Health and Care Excellence (NICE) is recommending evinacumab alongside diet and other low-density lipoprotein-cholesterol (LDL‑C) lowering therapies as an option for treating homozygous familial hypercholesterolaemia in people 12 years and over. Studies show that evinacumab can lower LDL‑C levels when statins and other lipid-lowering therapies have not reduced them enough.
Pregnancy and familial hypercholesterolaemia (FH)
Pregnancy can cause problems for women who have FH. This is because of 2 main reasons:
There is a normal increase in LDL cholesterol for all women during pregnancy.
The treatment options for FH are much more restricted during pregnancy, mainly because statins must not be taken when you're pregnant.
One type of cholesterol lowering medicine (called a bile acid sequestrant) is safe to take during pregnancy. Eating a low fat diet is also very important.
Another treatment called LDL-apheresis (a dialysis-like procedure that physically removes LDL-cholesterol from your blood stream) performed every 1-2 weeks has been successfully used for pregnant women with very high cholesterol levels.
If you have FH but your partner doesn't, then there is a 1 in 2 chance that your baby will also have FH. However, starting treatment for FH early in life can allow your child to lead a normal, healthy and full life.
Wie ist der Ausblick?
Wenn die Behandlung früh genug im Leben begonnen wird, haben Menschen mit FH die gleiche Lebenserwartung wie die Allgemeinbevölkerung.
The outlook (prognosis) for people with heterozygous FH is usually good if they maintain a healthy lifestyle, have regular checks and take their medication without fail. The most significant complication is heart disease or another cardiovascular disease that may develop at a younger age than usual.
Patientenauswahl für hohen Cholesterinspiegel

Herzgesundheit und Blutgefäße
Hyperlipidämie
Hyperlipidaemia means a high level of cholesterol or triglycerides in your blood.
von Dr. Toni Hazell, FRCGP

Herzgesundheit und Blutgefäße
hohen Cholesterinspiegel
Cholesterol is a fat chemical (lipid) that is made in the cells in your body. Many different cells make cholesterol but cells in the liver make about a quarter of the total. You need some cholesterol to keep healthy.
von Dr. Rosalyn Adleman, MRCGP
Häufig gestellte Fragen
What is primary hypercholesterolaemia?
The article focuses on familial hypercholesterolaemia (FH), which is an inherited condition resulting in high cholesterol. While the article doesn't use the term 'primary hypercholesterolaemia', FH falls under this category as it is a genetic cause of high cholesterol, distinct from high cholesterol caused by lifestyle factors or other medical conditions (which would be secondary hypercholesterolaemia).
How soon do children with FH typically need to start treatment?
Children with FH usually start treatment by the age of 10. However, if other family members have developed heart disease at a very young age, treatment might begin even earlier or involve more than one medication to lower cholesterol levels. In rare cases of homozygous FH, children might also need a treatment called apheresis.
What happens if a woman with FH becomes pregnant?
Pregnancy can present challenges for women with FH because cholesterol levels naturally increase, and many usual treatments, like statins, cannot be taken. A low-fat diet is important, and specific cholesterol-lowering medicines called bile acid sequestrants are considered safe during pregnancy. For very high cholesterol levels, a procedure called LDL-apheresis, which filters cholesterol from the blood, can be used every 1-2 weeks.
What kind of effect does early treatment have on the life expectancy of someone with FH?
If treatment for FH is initiated early in life and maintained consistently, individuals with the condition can expect to have a life expectancy similar to that of the general population. The outlook is generally good for those who adhere to a healthy lifestyle, attend regular check-ups, and take their medication as prescribed.
What does 'cascade testing' for FH involve?
Cascade testing means that if FH is confirmed in one person, all their first-degree relatives (parents, siblings, children) and second-degree relatives (uncles, aunts, nephews, nieces, grandparents, grandchildren) should also be referred for DNA testing. This is done at a specialist centre and is crucial for identifying other family members with FH so they can receive diagnosis and treatment, potentially preventing early heart attacks.
Weiterführende Literatur und Referenzen
- Lipid modification - cardiovascular risk assessment and the modification of blood lipids for the prevention of primary and secondary cardiovascular disease; NICE Clinical Guideline (July 2014 - updated May 2023)
- Ezetimib zur Behandlung von primärer heterozygoter familiärer und nicht-familiärer Hypercholesterinämie; NICE Technologie-Bewertungsleitlinie, Februar 2016
- Alirocumab zur Behandlung von primärer Hypercholesterinämie und gemischter Dyslipidämie; NICE Technologie-Bewertungsleitlinie, Juni 2016
- Evolocumab zur Behandlung von primärer Hypercholesterinämie und gemischter Dyslipidämie; NICE Technologie-Bewertungsleitlinie, Juni 2016
- Familiäre Hypercholesterinämie: Identifikation und Management; NICE Klinische Leitlinie (2008, zuletzt aktualisiert im Oktober 2019)
- Bempedoinsäure mit Ezetimib zur Behandlung von primärer Hypercholesterinämie oder gemischter Dyslipidämie; NICE Technologie-Bewertung, April 2021
- Inclisiran zur Behandlung von primärer Hypercholesterinämie oder gemischter Dyslipidämie; NICE Technologie-Bewertungsleitlinie, Oktober 2021
- Kardiovaskuläre Risikobewertung und Lipidmodifikation; NICE Qualitätsstandard, Mai 2023
- Kardiovaskuläre Erkrankungen: Risikobewertung und -reduktion, einschließlich Lipidmodifikation; NICE Klinische Leitlinie (Juli 2014 - zuletzt aktualisiert Mai 2023) Ersetzt durch NG238
Über den AutorVollständige Biografie anzeigen

Dr Colin Tidy, MRCGP
Allgemeinmediziner, Medizinischer Autor
MBBS, MRCGP, MRCP (Paediatrics), DCH
Dr. Colin Tidy ist ein NHS-Arzt mit Sitz in Oxfordshire.
Über den RezensentenVollständige Biografie anzeigen

Dr Krishna Vakharia, MRCGP
Chief Medical Officer für Gesundheit, Optum UK
MBChB, MRCGP(2013), BMedSci (hons), DFSRH, DRCOG, PGDipDerm (Distn)
Dr. Krishna Vakharia ist eine NHS-Hausärztin. Sie ist auch regelmäßige Prüferin für das postgraduale Diplom in Praktischer Dermatologie an der Cardiff University und zudem Chief Medical Officer für Gesundheit bei Optum UK.
Artikelverlauf
Die Informationen auf dieser Seite wurden von qualifizierten Klinikern verfasst und begutachtet.
Artikel auch verfügbar in Englisch, Deutsch, Spanisch, Französisch, Italienisch, Portugiesisch, Hindi, Hebräisch, Arabisch, und Schwedisch.
Nächste Überprüfung fällig: 13. Juni 2028
15. Juni 2023 | Neueste Version

Fragen, teilen, verbinden.
Durchsuchen Sie Diskussionen, stellen Sie Fragen und teilen Sie Erfahrungen zu Hunderten von Gesundheitsthemen.

Fühlen Sie sich unwohl?
Bewerten Sie Ihre Symptome online kostenlos
Abonnieren Sie den Patienten-Newsletter
Ihre wöchentliche Dosis klarer, vertrauenswürdiger Gesundheitsberatung - geschrieben, um Ihnen zu helfen, sich informiert, selbstbewusst und in Kontrolle zu fühlen.
Durch das Abonnieren akzeptieren Sie unsere Datenschutzrichtlinie. Sie können sich jederzeit abmelden. Wir verkaufen Ihre Daten niemals.