Glycogen storage disorders
Peer reviewed by Dr Helen Huins, MRCGPLast updated by Dr Roger Henderson, MBBSLast updated 22 Aug 2017
Meets Patient’s editorial guidelines
- DownloadDownload
- Share
- Language
- Discussion
- Audio Version
- Add to preferred sources on Google
This page has been archived.
It has not been reviewed recently and is not up to date. External links and references may no longer work.
Glycogen storage disorders are a group of inherited diseases. They result from a problem with one of the proteins (known as enzymes) involved in the conversion of glucose to glycogen, or the breakdown of glycogen back into glucose. They mostly tend to affect your liver and muscles. Most are diagnosed in childhood. Symptoms include weakness, tiredness and low blood sugar levels. Newer treatment possibilities provide hope for an improved outlook (prognosis). Most people with a glycogen storage disorder respond well to treatment. However, type II (infantile Pompe's disease) can be difficult to treat and can affect life expectancy.
At a glance
Glycogen storage disorders are a group of rare, inherited conditions affecting how your body uses glucose for energy.
They are caused by missing or faulty enzymes involved in converting glucose to glycogen or breaking glycogen back down.
There are over 12 types, mostly affecting the liver and muscles, but also other organs like the heart or kidneys.
Symptoms vary by type and can include low blood sugar, enlarged liver, muscle weakness, and developmental issues.
Diagnosis involves blood tests, scans, and sometimes a tissue biopsy.
Treatment aims to stabilise blood sugar and energy levels, often using corn flour or nutritional supplements.
The outlook depends on the specific type, but many people respond well to treatment.
What is glycogen?
Your body needs the sugar glucose to provide energy so that your cells and organs can function normally. Glucose is stored in your muscles and liver as a substance called glycogen. Glycogen is essentially made up of a number of glucose units (molecules) joined together. Various different proteins, known as enzymes, help to convert glucose into glycogen for energy storage. Other enzymes break down the glycogen back into glucose when more glucose is needed for energy.
Metabolism means all the chemical reactions in your body that are involved in keeping the cells in your body alive. Metabolism can be divided into:
Catabolism, which means the breakdown of substances in your body to make energy for your body to use.
Anabolism, which means making substances that are needed by the cells in your body.
What are glycogen storage disorders?
Glycogen storage disorders are a group of inherited diseases that result from a lack of, or abnormal functioning of, one of the proteins (enzymes) involved in the conversion of glucose to glycogen or the breakdown of glycogen back into glucose. Because there are a number of different enzymes involved in glycogen production and breakdown, there are a number of different glycogen storage disorders. In fact, there are over 12 types of glycogen storage disorder. Each disorder has a different enzyme lack or malfunction.
If the enzyme problem is with one of the enzymes involved in glycogen production (synthesis), this causes reduced amounts of normal glycogen to be produced and sometimes abnormal glycogen being produced.
If the enzyme problem is with one of the enzymes involved in glycogen breakdown back into glucose, this can lead to:
Low levels of glucose in your body (a condition known as hypoglycaemia).
A build-up of glycogen in your muscles and liver.
Because glycogen storage disorders are inherited and can affect energy production and metabolism within your body, they are also known as inborn errors of metabolism.
What are the different types of glycogen storage disorders?
Glycogen storage disorders are classified according to which protein (enzyme) is lacking or not working normally and also which part of the body is affected by the disease. Glycogen storage disorders mostly tend to affect your liver and muscles. However, some glycogen storage disorders can affect other parts of the body such as the kidney, heart, blood vessels, nervous system and bowel (see below). The different types of glycogen storage disorder include:
Type Ia (von Gierke's disease), type Ib.
Type II (Pompe's disease).
Type III (Forbes-Cori disease).
Type IV (Andersen's disease).
Type V (McArdle's disease).
Type VI (Hers' disease).
Type VII (Tarui's disease).
Type IX (liver phosphorylase kinase deficiency).
Type XI (Fanconi-Bickel syndrome).
Type 0 (Lewis' disease).
Type I glycogen storage disorder is the most common. About one quarter of people who have glycogen storage disorder have type I. It is due to a lack of the enzyme known as glucose-6-phosphatase. Type VIII and type X are now classified with type VI.
Who gets glycogen storage disorders?
Glycogen storage disorders are rare. It is estimated that about one baby in every 20,000 to 40,000 babies born has a glycogen storage disorder. Glycogen storage disorders are inherited. This means that they are passed on in families through your genes.
Autosomal recessive inheritance pattern
Most, but not all, glycogen storage disorders are inherited in a pattern called autosomal recessive inheritance.
Autosomal recessive pattern of inheritance
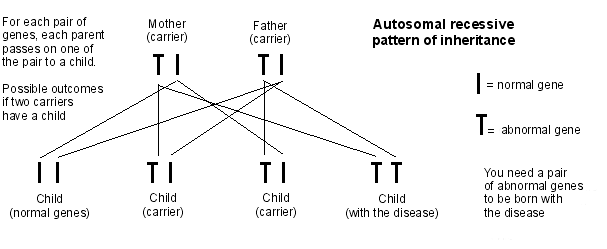
Someone can be a carrier for a glycogen storage disorder. This means that they are healthy but carry the faulty gene. For a child to be affected, both parents need to carry the faulty gene. If both parents are carriers of the faulty gene, the chance of each one of their children being affected by the glycogen storage disorder is 1 in 4.
X-linked inheritance pattern
This occurs in some people with glycogen storage disorder type IX.
X-linked inheritance pattern
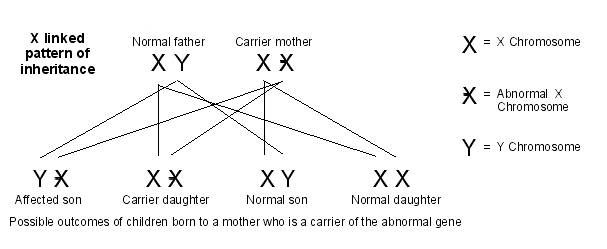
In this pattern of inheritance, females can be carriers for the disorder but the disorder only affects males. Females carry the faulty gene for the disorder on one of their two X chromosomes. The harmful effects of the gene are masked by their normal second X chromosome. However, as males have one X chromosome and one Y chromosome, if they have the faulty gene then the harmful effects are not masked.
What are the symptoms of glycogen storage disorders?
Some disorders affect the liver (types I, IV, VI and IX), some the muscles (types V and VII) and some both (type III). Disorders affecting the liver lead to liver enlargement and can be associated with a tendency to low blood sugar. Those affecting the muscles cause muscle weakness and sometimes kidney disease.
The symptoms and severity of symptoms vary between each type of glycogen storage disorder. The common features, particularly for type I, include:
Abnormally slow growth which may be detected by your child's health visitor or GP on routine examination.
Low blood sugar levels which can cause symptoms such as sweating, tremor, drowsiness, confusion and sometimes uncontrollable muscle movements (convulsions).
An enlarged liver which a doctor may feel when they examine your child's tummy (abdomen).
Noticeably weaker muscles. This can mean that your baby can appear floppy when they are held.
Being extremely fat (obesity).
Problems with bleeding and blood clotting.
Kidney problems.
In teenagers and adults, glycogen storage disorders usually cause tiredness (fatigue), feeling weak when exercising, or the feeling of aching and weak muscles.
Some glycogen storage disorders, particularly type Ib, can affect your immune system and make you more susceptible to infections.
Glycogen storage disorder type II can affect your heart, muscles, liver, nervous system and blood vessels. In babies, this can lead to breathing problems, muscle weakness and abnormal enlargement of the heart. This means that the heart cannot function normally which can lead to heart failure. In teenagers and adults, glycogen storage disorder type II can lead to muscle weakness in the limbs but also weakness of the muscles that are used to help your breathing. This can lead to difficulty breathing and respiratory failure.
How are glycogen storage disorders diagnosed?
Glycogen storage disorders can run in families. There may already be a history in your family of a glycogen storage disorder which may mean that your doctor suggests that you or your child should be tested. If your doctor suspects that you or your child may have a glycogen storage disease, they may suggest the following investigations:
Blood tests
These may include testing your blood sugar levels, which may be low, and also blood tests to check your kidney function and liver function. Blood tests for fats (lipids) and urate are important for diagnosing type I. A blood test to measure a substance called creatine kinase can give information about whether the glycogen storage disorder is affecting your muscles. Other blood tests may include checking your blood count to look for lack of iron in the blood (anaemia), which is very rare, and a test of your blood clotting.
Scans
A tummy scan (an abdominal ultrasound scan) can show if the glycogen storage disorder is causing enlargement of your liver. This is a painless test. It is the same type of scan that pregnant women have to look at the baby in the womb. Some gel is spread on to your abdomen. The ultrasonographer then moves the scanning probe over the surface of your abdomen. High-frequency sound waves allow them to produce an image of the inside of your abdomen that they can look at. Your doctor may also suggest an ultrasound scan of your heart, called an echocardiogram. This is because certain types of glycogen storage disorders can cause heart problems.
Biopsy
Your doctor may suggest that they take a sample of tissue (biopsy) either from one of your muscles or from your liver. However, liver biopsy is only rarely required because of improved gene testing.
The tissue sample will be sent to the laboratory for tests and an examination under the microscope. The levels of glycogen and fat in the tissue can be measured as well as the levels of proteins (enzymes) present. This can help to confirm the type of glycogen storage disorder.
Other tests
Other tests are sometimes carried out depending on the type of glycogen storage disorder that your doctor suspects.
What are the treatment options for glycogen storage disorders?
The treatment for glycogen storage disorders varies depending on which disorder you have. With most, the treatment aims are to stabilise your blood sugar and energy levels within your body. This is normally achieved by using corn flour (as used in thickening gravy) or nutritional supplements such as glucose. A high-protein diet may also be helpful in some glycogen storage disorders.
Some people with glycogen storage disorders who do not respond to nutritional supplements may occasionally need a liver transplant. People with glycogen storage disorders affecting their immune system may need to take regular antibiotic medication to protect against infection.
In some glycogen storage disorders, clinical trials have successfully used treatment involving replacement of the enzymes that are deficient or not working normally. For example, in glycogen storage disorder type II, this treatment has been shown to help reverse the heart problems and muscle weakness that can occur.
Other clinical trials are underway that are looking at gene therapy as a treatment. This is where doctors and scientists aim to cure genetic diseases by introducing normal genes into patients to overcome the effects of faulty genes, using techniques of genetic engineering.
What is the outlook for glycogen storage disorders?
The outlook (prognosis) depends on the type of glycogen storage disorder that you have. Most people with a glycogen storage disorder respond well to treatment. However, type II glycogen storage disorder (infantile Pompe's disease) can be difficult to treat and can affect life expectancy.
Because of the effects on muscles, your liver and your heart, you may have problems with breathing and heart function. This can sometimes lead to death which can occur at a very early age in some cases.
Your doctor will be able to discuss the outlook for a particular glycogen storage disorder in more detail. The possibilities of new treatments and therapies are constantly emerging. Enzyme replacement therapy and gene therapy mean that it is hoped that the outlook for glycogen storage disorders will soon be greatly improved.
Can glycogen storage disorders be prevented?
Glycogen storage disorders are hereditary conditions that run in families. If someone else in your family has a glycogen storage disorder (or if you personally have one, or one of your existing children has one), your doctor may refer you to a gene specialist (geneticist). The geneticist will be able to discuss the likelihood of your future child, or children, having a glycogen storage disorder.
When a woman becomes pregnant, there is also the possibility of having some tests carried out early in the pregnancy, to determine if the unborn baby has a glycogen storage disorder. The tests involve taking a sample of amniotic fluid from around the baby. This allows the doctor to study the levels of the proteins (enzymes) in the amniotic fluid and therefore determine whether the unborn baby has a glycogen storage disorder.
Patient picks for Genetic conditions

Children's health
Turner syndrome
Turner syndrome is a genetic condition that only affects girls. The most characteristic features of the syndrome are being short, having certain physical features (detailed below), and ovaries that do not work properly. Although there is no cure, there are treatments that can help most girls with Turner syndrome lead relatively normal lives.
by Dr Colin Tidy, MRCGP

Children's health
Sickle cell trait
If you inherit a gene from one of your parents, called a sickle cell gene, you have a condition called sickle cell trait. If you inherit a sickle cell gene from both parents, you get a double dose of the sickle cell gene. This causes a condition called sickle cell disease (SCD).
by Dr Sarah Jarvis
Frequently asked questions
What is the role of enzymes in my body concerning glycogen and glucose?
Enzymes are specific proteins in your body that play a crucial role in managing glucose (sugar) and glycogen. Some enzymes convert glucose into glycogen for storage, while others break down glycogen back into glucose when your body needs energy. A problem with any of these enzymes can lead to a glycogen storage disorder.
Are new treatments for glycogen storage disorders becoming available?
Yes, there is ongoing research and development for new treatments for glycogen storage disorders. Clinical trials have successfully used enzyme replacement therapy for some types, such as glycogen storage disorder type II, which has shown to help reverse heart problems and muscle weakness. Additionally, gene therapy is being explored, aiming to introduce normal genes to overcome the effects of faulty ones.
Can adults develop glycogen storage disorders, or are they only present from birth?
Glycogen storage disorders are inherited conditions, meaning they are present from birth due to faulty genes. However, the symptoms might not always be immediately apparent or severe in infancy. In teenagers and adults, these disorders commonly cause symptoms like tiredness, weakness during exercise, and aching or weak muscles.
How do doctors determine which specific type of glycogen storage disorder someone has?
Doctors classify glycogen storage disorders based on which specific enzyme is lacking or not functioning correctly, and which parts of the body are affected. They use various diagnostic methods including blood tests, scans, and sometimes a tissue biopsy to identify the exact enzyme deficiency and confirm the type of disorder.
If a glycogen storage disorder can affect the immune system, what does that mean for a patient?
If a glycogen storage disorder, particularly type Ib, affects the immune system, it means that the individual becomes more vulnerable to infections. To help protect against this, patients may need to take regular antibiotic medication.
Further reading and references
- Ozen H; Glycogen storage diseases: New perspectives. World J Gastroenterol. 2007 May 14;13(18):2541-53.
- Glycogen Storage Diseases, Type Ia, GSD1A; Online Mendelian Inheritance in Man (OMIM)
- Oldfors A, DiMauro S; New insights in the field of muscle glycogenoses. Curr Opin Neurol. 2013 Oct;26(5):544-53. doi: 10.1097/WCO.0b013e328364dbdc.
- Hicks J, Wartchow E, Mierau G; Glycogen storage diseases: a brief review and update on clinical features, genetic abnormalities, pathologic features, and treatment. Ultrastruct Pathol. 2011 Oct;35(5):183-96. doi: 10.3109/01913123.2011.601404.
About the authorView full bio

Dr Roger Henderson, MBBS
General Practitioner, Medical Author
MBBS, LMSSA
Dr Roger Henderson qualified as a GP in 1985 and has authored for EMIS since early 2013.
About the reviewerView full bio

Dr Helen Huins, MRCGP
General Practitioner, Medical Author
MB, BS, Lond, DCH, DRCOG, MRCGP, JCPTGP, DFFP
Helen qualified at Guy’s Hospital in 1989 and left London in 1990 to settle in the countryside.
Article history
The information on this page is written and peer reviewed by qualified clinicians.
Article also available in English, German, Spanish, French, Italian, Portuguese, Hindi, Hebrew, Arabic, and Swedish.
22 Aug 2017 | Latest version

Ask, share, connect.
Browse discussions, ask questions, and share experiences across hundreds of health topics.

Feeling unwell?
Assess your symptoms online for free
Sign up to the Patient newsletter
Your weekly dose of clear, trustworthy health advice - written to help you feel informed, confident and in control.
By subscribing you accept our Privacy Policy. You can unsubscribe at any time. We never sell your data.
More in children's health
- Anxiety in children
- Baby-led weaning
- Bedwetting medicine - desmopressin
- Bottle-feeding your baby
- Breastfeeding your baby
- Bronchiolitis
- Eczema in children
- Erythema toxicum neonatorum
- Feeding your baby
- Feeding your toddler
- Kawasaki disease
- Knock knees
- Meningococcal vaccine for meningitis
- Nausea and vomiting
- Necrotising enterocolitis
- Norovirus
- Retinopathy of prematurity
- Safeguarding children
- Turner syndrome